| Tabella 1 - Categorie di residui secondo la Direttiva 86/469 | |
| A) CATEGORIE COMUNI A TUTTI GLI STATI MEMBRI | |
| categoria I | a) Stilbene, derivati dello stilbene, loro sali ed esteri |
| b) Sostanze tireostatiche | |
| c) Altre sostanze ad azione estrogena, androgena o gestagena ad eccezione di quelle della categoria A, II | |
| categoria II | Sostanze autorizzate conformemente all'articolo 4 della direttiva 81/602/CEE ed all'articolo 2 della direttiva 85/649/CEE |
| categoria III | a) Sostanze inibenti - Antibiotici, sulfamidici ed altre sostanze antimicrobiche analoghe |
| b) Cloramfenicolo | |
| B) CATEGORIE SPECIFICHE | |
| categoria I - Altri farmaci | a) Sostanze impiegate contro endo- ed ectoparassiti |
| b) Tranquillanti e beta-bloccanti | |
| c) Altri farmaci veterinari | |
| categoria II - Altri residui | a) Contaminanti presenti negli alimenti del bestiame |
| b) Contaminanti presenti nell'ambiente | |
| c) Altre sostanze | |
| Tabella 2- Classificazione delle sostanze da ricercare come indicato nell'Allegato I della Direttiva 96/23/CEE | |
| Categoria A - Sostanze ad effetto anabolizzante e sostanze non autorizzate:
| |
| categoria A, 1 | stibeni, loro derivati e loro sali ed esteri |
| categoria A, 2 | agenti antitiroidei |
| categoria A, 3 | steroidi |
| categoria A, 4 | l attoni dell'acido resorcilico (compreso lo zeranolo) |
| categoria A, 5 | beta-agonisti |
| categoria A, 6 | sostanze incluse nell'allegato VI del regolamento (CEE) n. 2377/90 del Consiglio, del 26 giugno 1990 |
| Categoria B - Farmaci veterinari (3) e contaminanti ambientali: | |
| categoria B, 1 | sostanze antibatteriche, compresi sulfamidici e chinoloni |
| Categoria B, 2 - altri prodotti medicinali veterinari: | |
| B, 2a | antielmintici |
| B, 2b | coccidiostatici, compresi i nitroimidazoli |
| B, 2c | carbammati e piretroidi |
| B, 2d | tranquillanti |
| B, 2e | antinfiammatori non steroidei (AINS) |
| B, 2f | altre sostanze esercitanti un'attività farmacologia |
| Categoria B, 3 - altre sostanze e agenti contaminanti per l'ambiente: | |
| B, 3a | composti organoclorurati, compresi i PCB |
| B, 3b | composti organofosforati |
| B, 3c | elementi chimici |
| B, 3d | micotossine |
| B, 3e | coloranti |
| B, 3f | altri |
| Tabella 3 - Categoria di residui o di sostanze da ricercare a seconda del tipo di animali, loro alimenti e acqua di abbeveraggio e del tipo di prodotti animali di origine primaria come indicato nell'Allegato II della Direttiva 96/23/CEE | |||||||
| Tipo di animali Prodotti animali | Animali delle specie bovina, ovina, caprina, suina ed equina | Volatili da cortile | Animali d'acqua coltura | Latte | Uova | Carni di coniglio e di selvaggina selvatica Selvaggina da allevamento (*) | Miele |
| Categoria di sostanze | |||||||
| A, 1 | X | X | X | - | - | X | - |
| A, 2 | X | X | - | - | - | X | - |
| A, 3 | X | X | X | - | - | X | - |
| A, 4 | X | X | - | - | - | X | - |
| A, 5 | X | X | - | - | - | X | - |
| A, 6 | X | X | X | X | X | X | - |
| B, 1 | X | X | X | X | X | X | X |
| B, 2a | X | X | X | X | - | X | - |
| B, 2b | X | X | - | - | X | X | - |
| B, 2c | X | X | - | - | - | X | X |
| B, 2d | X | - | - | - | - | - | - |
| B, 2e | X | X | - | X | - | X | - |
| B, 2f | - | - | - | - | - | - | - |
| B, 3a | X | X | X | X | X | X | X |
| B, 3b | X | - | - | X | - | - | X |
| B, 3c | X | X | X | X | - | X | X |
| B, 3d | X | X | X | X | - | - | - |
| B, 3e | - | - | X | - | - | - | - |
| B, 3f | - | - | - | - | - | - | - |
| (*) La selvaggina selvatica è interessata solo per quanto concerne gli elementi chimici | |||||||
| Tabella 4 - I laboratori comunitari di riferimento (6) | ||
| LCR | Gruppo(A) | Sostanze |
| Rijksinstituut voor de Volksgezondheid en Milieu (RIVM) Bilthoven (Olanda) | A1 | Stilbene e suoi derivati compresi i loro sali ed esteri |
| A2 | Agenti antitiroidei | |
| A3 | Steroidi | |
| A4 | lattoni dell'acido resorcilico (compreso lo zeranolo) | |
| B 2 (d) | Tranquillanti | |
| B 3 (d) | Micotossine | |
| Agence Française de Sécurité Sanitaire des Aliments (AFSSA) Fougères (Francia)(B) | B1 | Sostanze antibatteriche compresi i sulfamidici e i chinoloni |
| B3 (e) | Coloranti | |
| Bundesinstitut für gesundheitlichen Verbraucherschutz und Veterinärmedizin (BgVV)Berlino (Germania) | A5 | beta-agonisti |
| B 2 (a) | Antielmintici | |
| B 2 (b) | Coccidiostatici compresi i nitroimidazoli | |
| B 2 (e) | antinfiammatori non steroidei | |
| Istituto Superiore di Sanità (ISS) Roma (Italia) | B 2 (c) | Carbammati e piretroidi |
| B 3 (a) | Sostanze organoclorurate (compresi i policlorobifenili, le policlorodibenzo-p-diossine ed i policlorodibenzofurani) | |
| B 3 (b) | Composti organofosforati | |
| B 3 (c) | Elementi chimici | |
| (A) Secondo la classificazione dell'Allegato I della Direttiva 96/23/CE; (B) Al LCR di Fougères competono anche i residui di Carbodox e Olaquindox | ||
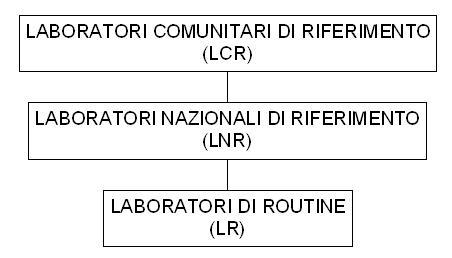
| Compiti dei LCR (7) | Compiti dei LNR (8) |
| 1. promuovere e coordinare la ricerca di nuovi metodi di analisi ed informare i laboratori nazionali di riferimento (LNR) circa i progressi compiuti nel settore dei metodi e dei materiali di analisi | 1. coordinare le attività dei laboratori nazionali abitualmente incaricati delle analisi dei residui ed, in particolare, di coordinare le norme ed i metodi d'analisi per ciascun residuo o gruppo di residui loro assegnati |
| 2. assistere i laboratori nazionali di riferimento per i residui al fine di porre in essere un sistema adeguato di garanzia della qualità basato sui principi di buona prassi di laboratorio (GPL) e sui criteri EN 45000 | 2. assistere l'autorità competente nell'organizzazio-ne del piano nazionale residui (PNR) |
| 3. approvare i metodi convalidati come metodi di riferimento da inserire in una apposita raccolta | 3. organizzare periodicamente prove comparative per ciascun residuo o gruppo di residui per i quali essi sono stati designati |
| 4. fornire ai laboratori nazionali di riferimento i metodi analitici di routine riconosciuti durante la procedura di fissazione di limiti massimi di residui | 4. garantire l'osservanza, da parte dei laboratori nazionali, dei limiti fissati |
| 5. fornire agli LNR i dettagli dei metodi analitici e le prove comparative da effettuare e comunicare loro i risultati di queste ultime | 5. garantire la diffusione delle informazioni fornite dai laboratori comunitari di riferimento |
| 6. fornire ai laboratori nazionali, che ne fanno richiesta, un parere tecnico sulle analisi delle sostanze per le quali sono stati designati come LCR | 6. assicurare al personale la possibilità di partecipare ai corsi di perfezionamento organizzati dalla Commissione o dai laboratori comunitari di riferimento |
| 7. organizzare esercizi interlaboratorio a beneficio degli LNR, con una frequenza determinata d'intesa con la CE. Ai fini di tali prove, gli LCR devono distribuire agli LNR campioni privi di analiti (i cosiddetti bianchi-campione) e campioni contenenti quantità note della sostanza da analizzare | |
| 8. identificare e la quantificare i residui nel caso in cui un risultato d'analisi dia luogo a contestazione tra due stati membri o tra UE ed un paese terzo | |
| 9. organizzare corsi di formazione e perfezionamento per gli esperti dei laboratori nazionali | |
| 10. apportare assistenza tecnica e scientifica alla Commissione, ivi incluso il programma delle norme, misure e prove | |
| 11. collaborare nel settore dei metodi e dei materiali d'analisi con gli LNR designati dai paesi terzi nel quadro dei piani di sorveglianza che devono essere presentati conformemente all'articolo 11 della Direttiva 96/23/CEE |
| Tabella 5 - Parametri da determinare nella validazione di un metodo in funzione del suo scopo | |||
| Tipo di metodo Parametri di Prestazione | Prove di identificazione | Determinazione di componenti principali | Determinazione di componenti minori |
| Esattezza/recupero | - | + | + |
| Precisione | - | + | + |
| Specificità | + | + | + |
| Limite di rivelabilità | - | - | + |
| Limite di quantificazione | - | - | + |
| Taratura | - | + | + |
| Campo di applicazione | - | + | + |
| Robustezza | + | + | + |
| Il segno "meno" significa che il parametro normalmente non viene stimato, mentre il segno "più" indica che il parametro normalmente viene stimato | |||
